Modern Approaches to Process Validation
Tuesday the 3rd January 2012
Validation has been a GMP requirement for all pharmaceutical companies for around 25 years now but, unfortunately, as an industry we have become focused on the bureaucracy of the validation process itself rather than the added value it can bring through assurance of product quality. Process validation is too often simply a compliance-driven, one-time event where three batches are manufactured and then
tested, around the time of filing a regulatory submission, which alone adds very little real value to the company or to patients. In addition, because our understanding of what really controls the quality of our products and processes has not always been sufficient, we have been unable to employ risk-based considerations. This generally means we have validated parameters that in reality may have little or no impact on product quality. In other words, we have created a perception of what is required for compliance and so in practice have done things which are unnecessary and simply added cost.
In November 2008 the FDA published a draft revision of their 1987 Guidance for Industry on Process Validation, which proposed a radical re-thinking of the whole concept of validation. This Guidance was made final in January 2011 and includes requirements for process design, the qualification of equipment and utilities and continued process verification after product launch. Significantly, the FDA requires this new process validation approach for existing marketed products, as well as new products.
In January 2010 the EU issued a concept paper that proposed a revision of the CHMP Note for Guidance on Process Validation along similar lines to the new FDA Guidance and it is understood that Annex 15 is also going to be revised.
The 2011 FDA Guidance contains a subtly different definition of process validation:
The collection and evaluation of data, from the process design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering quality product.
Thus process validation involves a series of activities taking place over the lifecycle of the product and process. In essence, it says that process validation starts with the product design and is a journey, not an event! This subtle but important change in philosophy underpins the whole of the document and presents a conceptual challenge to traditional pharmaceutical thinking. This new validation model is a science and risk-based approach and consistent with the ‘Quality by Design’ approach that is articulated in ICH Guidelines Q8, 9, 10 and 11.
The new Guidance states:
“A successful validation programme depends upon information and knowledge from product and process development. This knowledge and understanding is the basis for establishing an approach to control that is appropriate for the manufacturing process.”
Manufacturers should:
- Understand the sources of variation
- Detect the presence and degree of variation
- Understand the impact of variation on the process and ultimately on
product attributes
- Control the variation in a manner commensurate with the risk it
represents to the process and product.
It goes on to say:
Focusing on qualification efforts without understanding the manufacturing process may not lead to adequate assurance of quality.
Consequently, the Guidance recognises the following key stages in process validation.
The Stages of Process Validation
1. Process Design
Definition of the process based on knowledge gained
throughout development and scale-up activities
2. Process Qualification
Confirmation that the process design is capable of
reproducible commercial manufacture
3. Continued Process Verification
Ongoing assurance, gained during routine production, that
the process remains in a state of control
This three stage model is illustrated below:
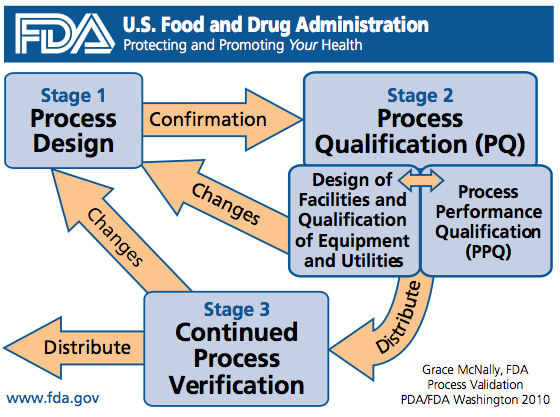
Stage 1 – Process Design
Process design involves two important phases:
- Building and capturing process knowledge and understanding
- Establishing a strategy for process control, which may or may not incorporate PAT principles
The process design stage delivers the planned commercial production and control records, which contain the operational limits and overall strategy for process control, which should be carried forward to the next stage for confirmation.
Stage 2 – Process Qualification
This has two elements:
- Design of the facility and qualification of the equipment and utilities. Reference is made to the science and risk-based ASTM E2500 standard on equipment and facility verification
- Process Performance Qualification (PPQ)
The new document states:
“The approach to PPQ should be based on sound science and the manufacturer’s overall level of product and process understanding.”
It goes on to say:
“It is not typically necessary to explore the entire operating range at commercial scale if assurance (of product quality) can be provided by other data.”
In other words, process definition is now part of process validation and, if the process is sufficiently understood and defined, then it may not be necessary to establish the ‘edges of failure’. It will, however, be necessary to define the ranges within which acceptable quality product is produced (a Design Space) and qualify these ranges.
This stage includes guidance on the writing and execution of PPQ protocols and reports.
Note: Stage 2 of the new FDA guide in practice covers the areas that were traditionally covered by DQ, IQ, OQ and PQ as referenced in the current EU Annex 15. The significance of the new FDA document is that it makes no reference to these terms and, in addition, it expects Stage 1 to have been completed before Stage 2 starts – i.e. the fundamentals of the product and process design must be understood before any qualification takes place.
Three is no longer the Magic Number
Although never mentioned in FDA Guidance, the principle of three consecutive acceptable batches has been implicit in FDA’s traditional expectations for process validation and is currently specified in EU GMP Annex 15. The 2011 FDA Guidance effectively consigns that approach to the dustbin of history.
The Guidance states “Each manufacturer should judge whether it has gained sufficient understanding to provide a high degree of assurance in its manufacturing process to justify commercial distribution of the product. Focusing exclusively on qualification efforts without also understanding the manufacturing process and associated variations may not lead to adequate assurance of quality”. It goes on to require that a clear conclusion be drawn as “to whether the data indicates the process met the conditions established in the protocol and whether the process is considered to be in a state of control. …This conclusion should be based on a documented justification for the approval of the process, and release of lots produced by it to the market in consideration of the entire compilation of knowledge and information gained from the design stage through the process qualification stage.”
Stage 3 – Continued Process Verification
Having confirmed that the process design is capable of reproducible commercial manufacture, the manufacturer must continually assure that the process remains in a state of control throughout commercial manufacture. Thus “An ongoing programme to collect and analyse product and process data that relate to product quality must be established”. The Guidance goes on to say that “The data should be statistically trended and reviewed by trained personnel. The information collected should verify that the quality attributes are being appropriately controlled throughout the process”.
Use of Statistics and Risk Assessment
The new FDA Guidance makes particular reference to the use of statistics and risk assessment, including references to ASTM standards on statistics, which should be used to:
- Understand sources of variation
- Identify critical variables
- Design a strategy for controlling critical variables
- Verify that a process is capable of consistently delivering quality product
- Continued monitoring throughout commercial production of the product
The new Guidance states “We recommend that a statistician or person with adequate training in statistical process control techniques develops the data collection plan and statistical methods and procedures used in measuring and evaluating process stability and process capability”.
This new approach to process validation encompasses equipment and utility qualification and is fully science and risk-based. It provides the pharmaceutical industry with the opportunity to re-think the whole concept of validation and ensure that these activities add real value to our businesses and to patients.